Bases de la Cromatografía Líquida
Definición y clasificación primaria de cromatografía
Según IUPAC (1-4), podemos definir el proceso como "un método físico de separación en el cual los componentes de la muestra a ser separados son distribuidos en dos fases, una de las cuales permanece inmóvil (fase estacionaria) mientras la otra (la fase móvil) se mueve en una dirección definida.
La Fase Estacionaria es una de las dos fases que componen el sistema cromatográfico. Puede ser un sólido, un gel o un líquido. Si fuera un líquido, puede estar distribuido sobre un sólido...
La Fase Móvil es un fluido que percola a través o a lo largo de un lecho estacionario en una dirección definida. Puede ser un líquido (cromatografía líquida) o un gas (cromatografía gaseosa)".
Según IUPAC, la cromatografía puede clasificarse de varias formas, según el método, forma del lecho estacionario, de la naturaleza física de la fase móvil y de acuerdo con el modo de separación (ver figura 1.1.).
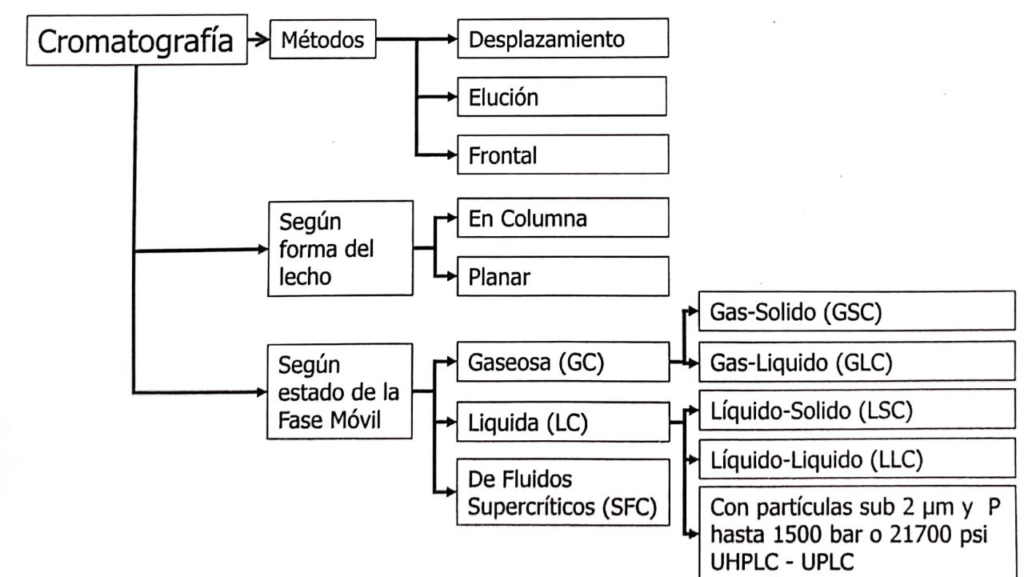
Figura 1.1.Clasificación de cromatografía según el método, forma física del lecho estacionario y estado físico de la fase móvil según IUPAC(1).
En relación con la cromatografía liquida (LC) en columna, podemos encontrar LC con partículas grandes operadas por gravedad o a baja presión, conocida como cromatografía liquida en columna, y LC con micropartículas que hacen necesario el uso de bombas para la elución de la fase móvil. Si las micropartículas son mayores de 2 μm es posible utilizar un sistema de HPLC, pero si tienen menos de 2 μm es necesario utilizar un sistema de UPLC o UHPLC.
La clasificación según el mecanismo de separación es algo más extensa y compleja, incluyendo las siguientes(1).
| Denominación del Método | Mecanismo de separación |
|---|---|
| Cromatografía de Adsorción | Diferencia de afinidad de adsorción entre componentes de la muestra y los sitios activos de la fase estacionaria |
| Cromatografia de Afinidad | Especificidad biológica de la interacción analito-ligando |
| Cromatografía en Contraco- rriente(CCC) | Partición entre dos fases liquidas inmiscibles sin soporte sólido |
| Cromatografía Quiral o Enan- tioselectiva | Separación por interacción con un se lector quiral del sistema, tipicamente la fase estacionaria pero también puede estar en la fase móvil |
| Cromatografia de Exclusión por tamaño(SEC) | Separación por exclusión (tamaño y/o forma del analito) con curvas de calibración basadas en el peso molecular. Según la fase móvil puede ser GFC(acuosas) o GPC(orgánicas) |
| Cromatografía Hidrodinámica | Separación de macromoléculas por diferencia de velocidad de fase móvil entre el eje y las paredes de la columna |
| Cromatografía de Interacción Hidrofílica-HILIC | Partición/Intercambio iónico en columna de fase normal, emplea una fase móvil orgánica con pequeña proporción de agua |
| Cromatografía de Interacción Hidrofóbica-HIC | Separación por interacción entre sitios hidrofóbicos de la fase estacionaria y sectores hidrofóbicos de las proteínas que se exponen por efecto de la fuerza iónica |
| Cromatografía de Intercambio Iónico | Separación por intercambio iónico entre grupos funcionales de la columna y componentes de la muestra. El intercambio puede ser de aniones o de cationes |
| Cromatografía de Exclusión Iónica | Exclusión de la partícula y elución tem prana de componentes iónicos, mientras compuestos de carga opuesta se retienen y eluyen después |
| Cromatografía de Apareamiento Iónico | Partición de pares iónicos formados entre el analito iónico y un aditivo de carga opuesta, en un buffer apropiado |
| Cromatografía de Intercambio de Ligandos | Separación basada en la capacidad del analito de formar complejos con metales |
| Cromatografía Micelar | Cromatografía en fase reversa donde la fase móvil contiene un surfactante en concentración superior a la micelar critica |
| Cromatografía en Fase Normal | Separación donde la fase estacionaria es más polar que la fase móvil |
| Cromatografia de Partición | Separación basada en la diferencia de solubilidad de los componentes de la muestra entre la fase móvil y la fase estacionaria |
| Cromatografía en Fase Reversa | Separación de LC donde la fase móvil es más polar que la fase estacionaria |
HPLC - UPLC - UHPLC
HPLC proviene de las siglas en inglés de High Performance Liquid Chromatography, que será aquí traducido como Cromatografía Líquida de Alta Performance y manteniendo aquel acrónimo. Se prefiere esta denominación a otras utilizadas en otros medios como CLAP (castellanización de las siglas en inglés), de alto desempeño o de alta eficacia, también utilizado por otras fuentes. Tampoco se traducirá como Cromatografía Liquida de Alta Presión como reflejo de las siglas utilizadas por primera vez por Csaba Horvath, pionero de esta ciencia, por el simple motivo que la presión no es una herramienta de separación sino la consecuencia natural de forzar el pasaje de un líquido a través de una columna rellena con macropartículas. Un hecho más claro pero que ha llevado a ciertos malentendidos es la llegada de UHPLC. Dado que los sistemas de HPLC "convencionales" habían llegado a dispensar presiones de hasta 6000 psi, ese nivel de presión se convirtió en un estándar de la industria. Sin embargo, la evolución de la partícula cromatografía a diámetros menores de 2 μm hizo necesario superar esa barrera. El primero en hacerlo fue Waters Corp., quien llamó al sistema UPLC® por "cromatografía liquida de ultra performance" y registró el nombre y las siglas, evitando el uso de este acrónimo por otras empresas de instrumental.
Estos sistemas pueden hoy llegar a presiones de hasta 1500 atm o casi 22000 psi, y es claro que el objetivo de su creación fue simplemente utilizar la partícula sub-2 creada, para lo cual era necesario aumentar la presión máxima de trabajo y disminuir tanto como fuera posible el volumen muerto del sistema, reducir el volumen de celda y aumentar la velocidad de respuesta del detector, factores imprescindibles para no perder la eficiencia ganada con la reducción del tamaño de partícula.
Al conseguir trabajar a mayor presión, con partículas de menor diámetro y por tanto más eficientes, parece haberse interpretado que esta moda-lidad era mejor, y por tanto diferente a HPLC y digno de otro nombre. Ante la novedad, la competencia en desventaja comercial en principio se opuso a esa modalidad argumentando falta de mérito y riesgos físicos para trabajar a presiones tan altas, reforzando la idea de que se trataba de otro sistema. Sin embargo y con la difusión creciente de la modalidad, y ante la mayor velocidad de trabajo, mayor eficiencia y menor consumo de solvente, finalmente comenzó el desarrollo y producción general de estos instrumentos, que se llamaron UHPLC para evitar el uso de un nombre ya registrado.
Sin embargo, se había producido una mella en la confianza de algunos usuarios, la sensación de que realmente podía ser otro método, y la consecuencia fue la imposibilidad, en ámbitos regulados, por ejemplo, USP, EP, JP, de transferir procedimientos desarrollados y validados en HPLC a UHPLC, simplemente porque se "trataría" de otro modo de separación. Sin embargo, las columnas, rellenos y funcionalidad de los rellenos es la misma, la fase móvil es la misma, los mecanismos de separación, la interpretación de los cromatogramas y los cálculos son los mismos. Fue necesario esperar la maduración del sistema para que fuera aceptado, y que se comprobara experimentalmente la equivalen cia, para que fuera posible la transferencia de procedimientos, lo cual se tradujo en el primer documento armonizado del capítulo de cromato-grafía de la USP (5), EP y JP en 2017, donde se propuso la transferencia de métodos de HPLC a UHPLC y viceversa en entornos regulados. Esto no quita por supuesto, que los métodos oficiales deben ser verificados antes de su implementación según prescriben la USP (6), FDA (7),ISO(8) y AOAC (9), ya sea que hayan sido transferidos a UHPLC o no. El concepto más importante es la conclusión de que ambas modalidades son equivalentes, y que los procedimientos oficiales pueden ser escalados, verificados y utilizados tal como fueron validados originalmente, sin pérdida del estatus "oficial".
El mecanismo de la separación
Volviendo a la definición de IUPAC, “Cromatografía es un método físico de separación en el cual los componentes de la muestra a ser separa-dos son distribuidos en dos fases...".
De modo que debemos evaluar un sistema de tres componentes, la muestra, la fase estacionaria y la fase móvil. En la figura 1.2, la muestra tiene tres componentes, cada uno de los cuales tiene una estructura química diferente, caracterizada por la presencia de grupos funcionales que la identifican, por ejemplo, grupos hidroxilo fenólico o alcohólico, aminos primarios o secundarios, aldehídos, cetonas, etc. A su vez, la fase móvil y la fase estacionaria tienen sus propios grupos funcionales, que le otorgan afinidad por grupos funcionales complementarios (ver figura 1.1).

Figura 1.2. Componentes de la muestra y su interacción con el sistema, fase móvil y fase estacionaria. La funcionalidad química de cada componente determina su afinidad por los estos componentes, y de ese modo, su mayor o menor retención en la columna. Se esquema-tizan tres tipos de moléculas, un hidrocarburo lineal, una molécula dipolar y un fenol como molécula polar.
Existen diversas interacciones que modelan la afinidad entre molécu-las. Una de las más importantes es el puente de hidrogeno, donde un átomo de hidrogeno está unido covalentemente a un átomo electronegativo en una molécula donante de protones que lo expone. En el entorno puede encontrar una molécula aceptora de protones con otro átomo electronegativo, ávida por ese protón a quien atrae, pero no puede tomarlo por estar unido covalentemente a primera molécula. Esta interacción está muy difundida en la naturaleza, por ejemplo, entre grupos carbonilos y aminos de las proteínas para formar su estructura tridimensional, entre bases complementarias del DNA, entre el agua y el hidroxilo de un alcohol, o un carbonilo, etc.
Otras interacciones incluyen las electrostáticas entre aniones y cationes, las interacciones hidrofóbicas e hidrofílicas que hacen que no se mezcle aceite y agua que se agrupan entre sí, las interacciones estéricas que dificultan el ingreso de una molécula en la fase ligada por impedimento estérico, debido simplemente al tamaño del analito, las fuerzas de London, débiles y producidas por los dipolos temporarios que se crean en cada átomo por movimiento de sus electrones, las interacciones dipolares producidas por moléculas de polaridad permanente como el acetonitrilo entre otras fuerzas de retención (figura 1.3).
En la separación cromatográfica (figura 1.4), la fase estacionaria contenida en una columna es equilibrada con un caudal constante defase móvil. La salida de la columna se conecta a un dispositivo de monitoreo que mide alguna propiedad fisicoquímica del líquido que eluye, en el caso de la figura un detector fotométrico o espectrofotométrico, pero puede ser de conductividad, fluorescencia, amperométricos, etc.
Una vez equilibrado el sistema se inyecta la muestra constituida por una solución que contiene varios componentes, los cuales viajan a través de la columna transportados por la fase móvil. De acuerdo con la funcionalidad química, cada uno de los componentes mostrará mayor afinidad por la fase móvil y eluirá más rápidamente, o mayor afinidad por la fase estacionaria y se retendrá según esa interacción.
Como resultado,los componentes viajarán a través de la columna con diferente velocidad y serán separados. La fase móvil eluída pasa a través de una celda de flujo donde es monitoreada porel sistema apropiado, en este caso un haz de luz que atraviesa la celda de flujo para encontrar una fotocélula, sensible al cambio de transmisión/absorción de la luz. La fase móvil continúa su camino y la señal de la fotocélula, proporcional a la absorción instantánea del eluido que atraviesa la celda, se transmite a un registrador grafico o a un sistema de registro y procesamiento de datos en una computadora. El resultado es un cromatograma, representación de la señal generada por la fotocélula en función del tiempo (ver figuras 1.7 a y b).
El cromatograma muestra entonces la separación de los componentes de la solución inyectada en un gráfico de la señal adquirida por el detector (mV) en función del tiempo (minutos). El tiempo de retención (tomado como el tiempo de elución de vértice del pico), es consecuencia de la estructura química y funcionalidad, y es por tanto función primaria de la identidad del componente. El tiempo de retención de A identifica primariamente al compuesto A como componente de la solución inyectada, al igual que B y C. Por su parte, la intensidad de la señal, medida como altura de pico o de área bajo la curva es función primaria de la concentración.
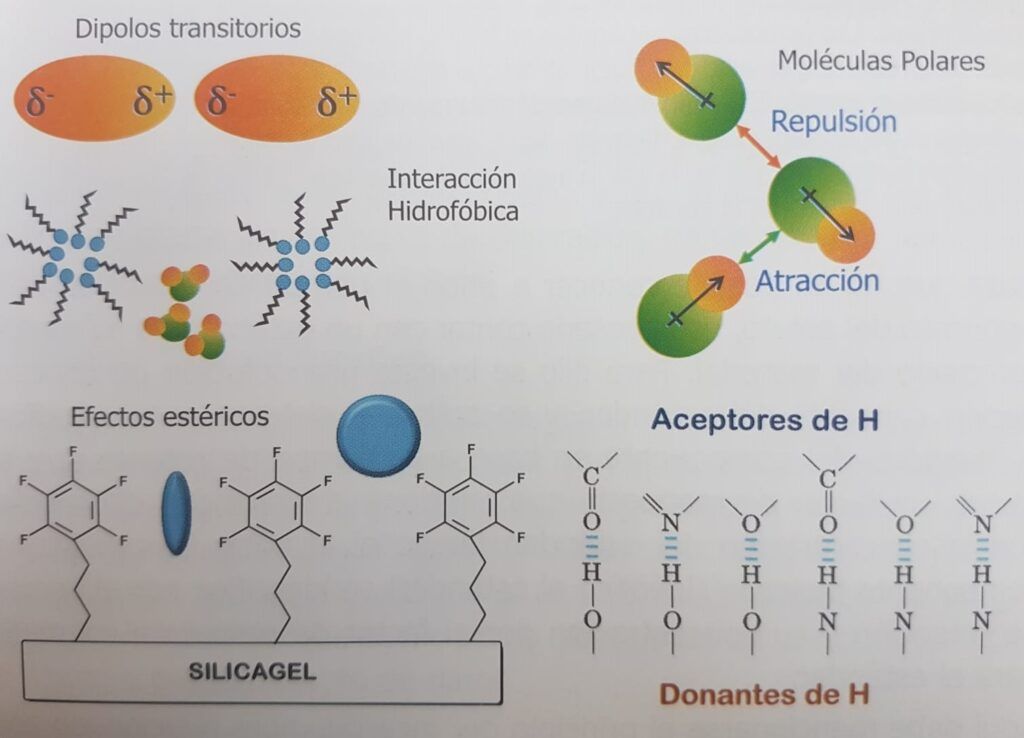
Figura 1.3. Algunos ejemplos de interacción entre el analito, la fase móvil y la fase estacio-naria: dipolos transitorios de London, interacción por dipolos permanentes, efectos estéricos, interacción hidrofóbica e hidrofílica y puente de hidrógeno.

Figura 1.4. Representación esquemática de la separación cromatográfica y su resultado, el cromatograma. Los compuestos separados en la columna son monitoreados a su salida por medio de un detector, en este caso fotométrico que registra la señal, en este caso absorción, en función del tiempo. El volumen de elución/tiempo de retención es función primaria de la identidad del analito, ya que depende de su funcionalidad química. La intensidad de la señal representada por altura de pico o área bajo la curva son función de la concentración de cada analito eluido de la columna.
Dado que no es posible conocer a priori el tiempo de retención ni la respuesta del soluto, es necesario contar con un estándar de referencia apropiado del material. Para ello se inyecta una solución de concen-tración conocida delIestándar, y se calibra el sistema para adjudicar la identidad del componente en base a su tiempo de retención, y se calcula un factor de respuesta que relaciona la intensidad de la señal con la concentración del estándar. Luego al inyectar la muestra, el componente buscado (idéntico al estándar) se identifica por el tiempo de retención y su concentración por el factor de respuesta estimado para el estándar.
Aquí debe mencionarse el principio der incertidumbre relacionado con la identidad. Si bien es claro que la identidad de los componentes A, B y C es función del tiempo de retención, es también claro que en la naturaleza seguramente existen decenas o cientos de compuestos que eluirán con un tiempo de retención similar o indiferenciable del tiempo de retención del componente buscado. Para el caso de la figura, pueden existir cientos de compuestos que eluyen en 10 minutos de corrida. Entonces, la validez de la primera aseveración, identidad, será más confiable en los ámbitos donde la variabilidad de componentes no sea muy alta, por ejemplo, en una industria que maneja unas pocas decenas de materiales para producir unos pocos productos. A medida que la diversidad se expande, va siendo necesario contar con herramientas de confirmación de identidad, por ejemplo, por análisis estructural (análisis espectroscópico simultáneo, análisis de pureza de pico, peso molecular) para mantener control sobre la validez del resultado. Y habrá casos en los que la diversidad es absolutamente compleja, como en el caso de pruebas bioanalíticas donde las matrices biológicas aportan un número muy elevado de interferentes potenciales, o en un screening medioambiental, o en el análisis de alguna estructura en matrices naturalmente complejas como frutas o extractos vegetales. En estos casos puede ser absolutamente necesario contar con detectores muy selectivos, como acoplamientos masa-masa o de tiempo de vuelo.
En relación con la incertidumbre relacionada con la concentración, es claro que debe buscarse una correlación entre la respuesta y la concentración del estándar en el sistema usado, y que la respuesta del analito debe interpolarse en esa curva de calibración, en un sistema calificado, usando un procedimiento validado.
El Sistema Cromatrográfico
La figura 1.5 muestra esquemáticamente la versión más simple de un sistema de HPLC. Es necesario contar con al menos un reservorio de fase móvil, una bomba, inyector, columna y detector, con los tubos y uniones correspondientes. El detector debe conectarse mínimamente a un registrador gráfico XY, aunque el análisis moderno estimula el uso de una computadora que actúa como sistema de adquisición, almacenamiento y procesamiento de datos.

Esquema básico de un cromatógrafo. Los componentes mínimos imprescindibles son el reservorio de fase móvil, la bomba, el inyector, la columna, el detector y un recipiente de desechos, todos estos unidos por tuberías de diámetro interno apropiado. La señal eléctrica en función del tiempo es tomada por un dispositivo que varía desde un registrador grafico hasta una computadora, y en el último caso, la posibilidad agregada de almacenar la señal para reprocesamiento o control regulatorio.
Si bien ese esquema básico puede ser suficiente para gran número de aplicaciones, es inevitable su expansión hacia sistemas más complejos por necesidad de optimización y automatización (ver figura 1.6). Así, por ejemplo, la confiabilidad del análisis instrumental recae en gran medida en el análisis estadístico, que promueve el procesamiento de replicados para calcular la incertidumbre.Esto lleva a la conveniencia de inyectores automáticos, que por otra parte permiten la optimización de tiempo y recursos.
Luego, en ocasiones es necesario utilizar gradientes de elución, lo que permite el uso de múltiples solventes para preparar in situ la fase móvil, los cuales pasan por desgasificadores on-line antes de llegar a la bomba. El gradiente puede formarse utilizando una o dos bombas.
Luego, es posible y en ocasiones conveniente usar un horno de columna para alojar una o más de una columna cromatográfica. El sistema puede programarse para usar la primera, lavarla, acondicionar la segunda y luego usarla y lavarla y apagar el sistema al terminar, todo sin necesidad de la operación del analista. La diversidad de analitos puede hacer necesario el uso de más de un detector, y el eluido de la columna no necesariamente debe descartarse, sino que los componentes separados pueden ser recolectados por sistemas apropiados.

El esquema básico de la figura 1.5 sumará complejidad en trabajos de desarrollo y de rutina, básicamente para mejor aprovechamiento de la automatización. Es frecuente dejar que el sistema prepare la fase móvil en lugar de hacerlo en mesada. Eso demanda el uso simultáneo de más de un solvente, que también puede ser desgasificado in situ. Tampoco es infrecuente utilizar más de una columna, o más de un detector. O separar las fracciones en modo preparativo en lugar de descartar las fracciones separadas. Y lavar las columnas antes de cambiar a la siguiente, todo en forma automática.
| Cromatograma | Prueba de aptitud del sistema | |
| Material | Solución estándar 1019 | Origen: Laboratorio QA |
| Analista | FQ-SR | Vial: 1 |
| Fecha | 26/07/2022 | Hora: 12:19:46 |



Breve reseña histórica de la Cromatografía Líquida
Lo que sigue son pinceladas del desarrollo histórico de la cromatografía líquida y HPLC que fueron tomadas de artículos publicados por renombrados investigadores de la cromatografía. No pretende ser una descripción exhaustiva o completa del desarrollo de la cromatografía y seguramente es injusta con muchos investigadores cuya contribución fue fundamental para esta técnica esencial en prácticamente todas las ciencias modernas, pero su lectura puede ayudar a comprender los mecanismos, y tal vez a mejorar los procesos actuales. Muchos de los acontecimientos citados pueden mostrar brevemente el origen y algunos hitos que nos permitieron llegar aquí(11-17).
El inicio no puede excluir la invención de la cromatografía, que debemos a Mikhail Semyonovich Tsweet quien la desarrolló en 1900. Tsweet nació en 1872 en Asti, Italia, hijo de madre italiana y de un oficial ruso. Muy joven se mudó a Suiza donde se graduó y se dedicó a la botánica. Luego siguió a su padre cuando éste fue llamado nuevamente a Rusia y se radico en San Petersburgo. Debió revalidar sus títulos en Moscú, donde ejerció actividades académicas, así como en Varsovia y en Lituania. Usó por primera vez el nombre de Cromatografía en sus publicaciones de 1906 (ver figura 1.8). En suus trabajos llenaba tubos de vidrio con un sólido, en el caso de la figura con carbonato de calcio y lo equilibraba con un líquido, disulfuro de carbono. Luego de introducir clorofilas en solución en el extremo superior, eluía el líquido a través de la columna con lo cual los pigmentos se separaban en la columna y dejaban ver una secuencia de bandas coloreadas. En ese momento creó el nombre de cromatografia, del griego chroma (color) y grapho (escritura), o sea, escritura en color.

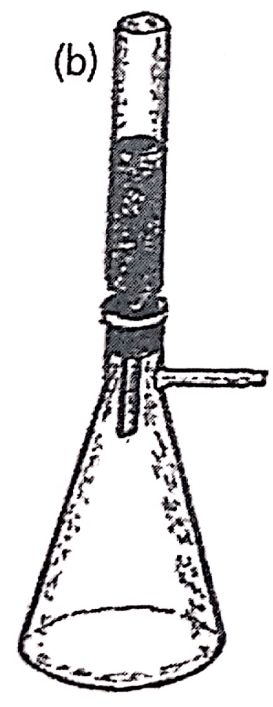

Figura 1.8. Dibujos del artículo publicado en 1906 por Tsweet. Incluye en (a) un aparato con cinco columnas simultaneas con manómetro de mercurio y una pera de goma para dispensar la fase móvil. También muestra en (b) una columna tipo embudo de 2-3 mm de diámetro y 20-30 cm de longitud montada sobre un Kitasato y en (c) una separación cromatográfica de pigmentos, utilizando carbonato de carbono como fase estacionaria y disulfuro de carbono como fase móvil.Reproducido con autorización de LC-GC.
En realidad, no se hacía eluir de la columna a los componentes de la muestra tal como hacemos actualmente, sino que constituían dentro de ésta un patrón de colores que representaba la composición de las clorofilas. El método tuvo gran repercusión,pero naturalmente tenía grandes limitaciones al no poder visualizar componentes incoloros, y al no permitir la elución de los componentes de la columna.
La cromatografía en capa delgada (TLC) presentó una solución a este problema.La TLC podía considerarse como una cromatografía en columna abierta, de modo que, al acceder al material, podían efectuarse revelados específicos o universales según fuera necesario. Por ejemplo, la aplicación de un aerosol de ninhidrina seguido de calor revela los aminoácidos,incoloros, que toman color azul. Los vapores de iodo,al exponer la placa seca luego de la corrida a sus vapores, se fijan sobre los compuestos orgánicos con dobles o triples enlaces confiriéndoles color marrón rojizo. Y la simple aspersión de ácido sulfúrico seguida por calentamiento en estufa a 100°℃ carboniza los compuestos orgánicos que se visualizan como manchas oscuras sobre la placa blanca.
HPTLC de aceite de chamomile, distintos revelados
UV 254 nm con y sin filtro
UV 366 nm Post derivatización
Derivatización anisaldenido Reflactancia R+T Transm.

Figura 1.9.Cromatografía en placa delgada de aceite de chamomile mostrando el resultado del empleo de diversos reveladores para la visualización de los compuestos separados.
Uno de los primeros precursores de esta técnica fue Edgar Lederer y tuvo avances más recientes con Egon Stahl. El impacto de la TLC fue inmediato, y debido a su simplicidad y bajo costo gano rápidamente terreno en el campo de la química analítica. En el momento de la mayor difusión de la cromatografía liquida y gaseosa, la TLC siguió desarrollándose en Europa del este. Hoy se sigue utilizando, aunque con mucho menor difusión por el avance de HPLC.
Poco después, la cromatografía liquida (LC) tuvo un avance muy significativo en la década del '40 cuando Archer J.P. Martin y Richard L.M. Synge crearon la cromatografía de partición o cromatografía liquido-liquido (LLC), opuesta a la cromatografía líquido sólido (LSC) que predominaba en ese momento, y tal fue el impacto de ese desarrollo que por el recibieron el premio nobel de química de 1952.
Martin investigaba la vitamina E en Dunn Nutritional Laboratory, y estaba experimentando con la separación de carotenos fases heterogéneas. Había construido para ello un sistema de 45 tubos encadenados que servían como ampollas de decantación, separados por 90 válvulas que impedían el retorno del líquido a la ampolla anterior. El esquema de su aparato no fue publicado.
Synge por su parte investigaba los aminoácidos de la lana en IWS, y el método empleado consistía en acetilar los aminoácidos para extraerlos en cloroformo. Comprobó con entusiasmo la diferencia en los coeficientes de partición cloroformo-agua de los aminoácidos acetilados, lo cual presentaba perspectivas promisorias para la extracción líquido-líquido,y para esto fue contactado con Martin, de gran experiencia en la distribución en contracorriente que había desarrollado un aparato que lo había hecho conocido en Cambridge. Poco tiempo después se encontraron trabajando en la Wool Research Association en Leeds, UK, donde debieron crear otro aparato porque el que tenían no era compatible con el sistema agua-cloroformo. El nuevo aparato de flujo continuo tenía 39 "platos teóricos" (la figura 1.10 representa un sistema ideado por Lyman Craig en los '40 (18) no necesariamente idéntico al sistema de Martin, pero que muestra un tren de extracción en contracorriente utilizado en aquellos años).
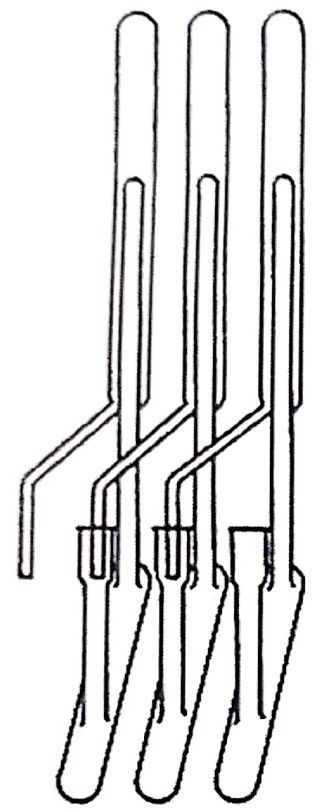

Figura 1.10. Tren de distribución en contracorriente desarrollado por Lyman Craig en 1944, que simplificaba notablemente el proceso de extracción múltiple en ampollas de decantación. Publicado con autorización de American Chemical Society(18).
Según se indicaba, era un sistema complicado, que debía ser atendido por dos operadores en ciclos de 4 horas, adormecidos por los vapores de cloroformo. Los resultados no fueron demasiado satisfactorios, y Martin y Synge trataron de mejorar el sistema creando variantes. Un sistema intermedio era un tubo de vidrio con las fibras de lana y algodón orientadas en el sentido del tubo, con el cloroformo moviéndose en un sentido y el agua en el opuesto. Tampoco funcionó como se esperaba hasta que finalmente Martin pensó que no era necesario mover los dos líquidos, que podía dejarse uno estacionario en una columna y mover el otro.
Para lograr este propósito impregnaron con agua la silicagel que usaban como desecante en la balanza y con este material empacaron una columna de vidrio, agregaron la mezcla de aminoácidos acetilados y vertieron cloroformo en la entrada, tal vez como en el esquema de la figura 1.11). El agregado de naranja de metilo les permitía seguir la trayectoria de los aminoácidos a través de la columna. Hoy puede sonar muy simple, pero sin duda fue un arduo trabajo preparar apropiadamente la fase estacionaria (agua) sobre un soporte que no se había fabricado para eso (silicagel) y preparar apropiadamente la fase móvil (cloroformo) en forma reproducible. Martin desarrollo en paralelo la teoría de la cromatografía basada en conceptos de destilación, y el trabajo fue enviado en dos partes en noviembre de 1941 al Biochemical Journal, y publicado en diciembre de 1941 como Una nueva forma de cromatografía empleando dos fases liquidas (19-20) En base a este trabajo Martin y Synge recibieron en 1952 el Premio Nobel de Química por la Invención de la Cromatografía de Partición.
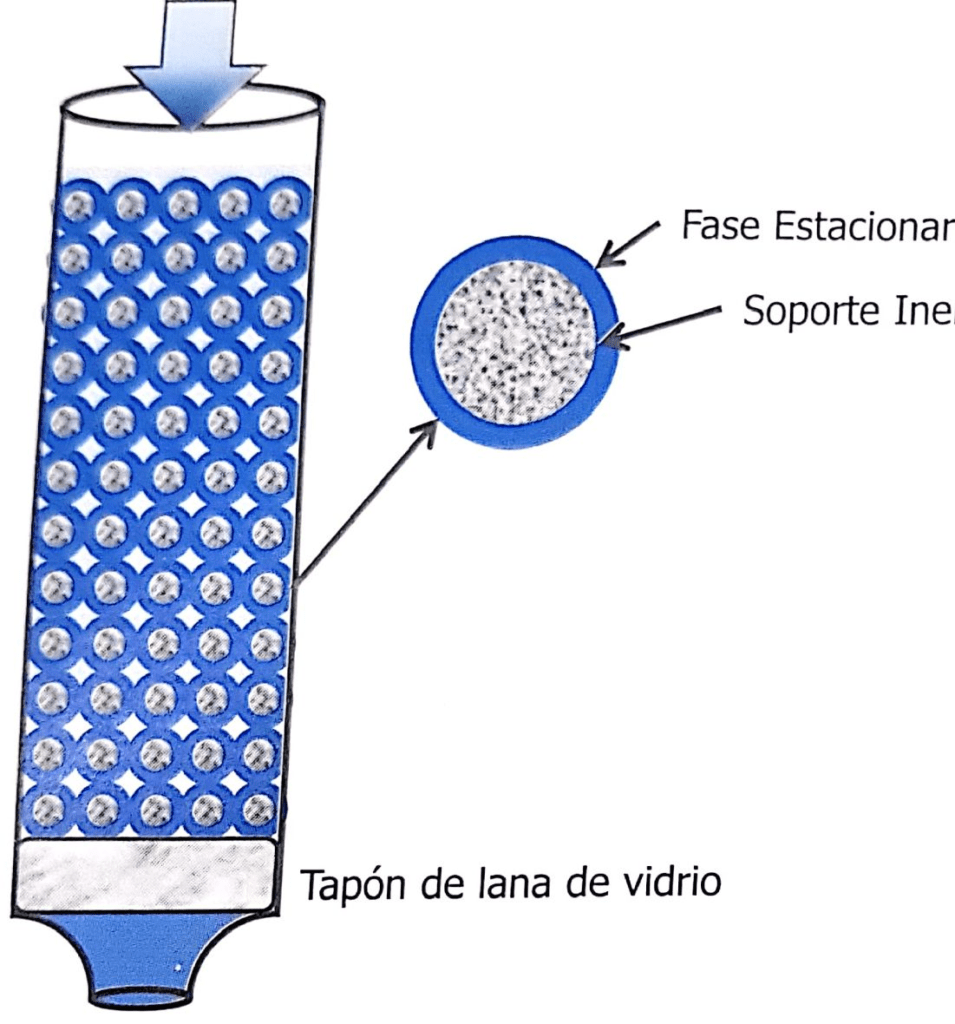
Figura 1.11. Esquema de la co-lumna de cromatografía de par-tición o LLC. La partícula solida se comporta como un soporte inerte,y la fase estacionaria es el líquido que la embebe,y que es inmiscible con la fase móvil, y con el solvente de la muestra.
Poco tiempo después Synge se apartó del grupo para investigar aminoácidos en antibióticos, y fue el primero en elucidar la secuencia en un polipéptido usando cromatografía en papel.
Por su parte Martin se unió en 1948 al Medical Research Council y se ocupó del análisis de ácidos grasos por cromatografía. Como su sistema tenía una fase estacionaria polar y una fase móvil poco polar (agua y cloroformo), los coeficientes de partición no lo favorecían, de modo que decidió desarrollar una fase estacionaria hidrofóbica para retener los ácidos grasos. Lo consiguió junto a Howard (20), tratando kieselguhr (tierra de diatomeas) con Dimetil diclorosilano (ver figura 1.12).
A diferencia de la cromatografía de partición líquido-líquido desarrollada en el '41 donde la fijación del líquido a la fase estacionaria era una simple retención mecánica en los poros y por lo tanto inestable, esta reacción unía la molécula silanizante con el soporte por unión covalente, dejando el líquido no polar (el dimetilsilano) como la fase estacionaria hidrofóbica, inmiscible con el solvente polar de la fase móvil. Martin usó este sistema para separar ácido láurico de esteárico por partición.

Figura 1.12. Reacción de silanizacion con la que Martin y Howard consiguieron unir covalentemente un líquido, Dimetil diclorosilano al soporte inerte de LLC, kieselguhr o tierra de diatomeas.
Por otra parte, Martin había predicho el uso de una fase móvil gaseosa en 1941, pero no desarrolló la idea hasta 1950 cuando llevó la fase estacionaria silanizada a la GC, publicando el método como cromatografía de partición gas-líquido en octubre de 1950. Este método demostró su utilidad en poco tiempo y fue base del desarrollo de la cromatografía gas-líquido (GLC).
En poco tiempo este material volvió a migrar a cromatografía liquida y fue usada por primera vez en 1967 por Aue y Hasting en la Universidad Dalhouise en Nova Scotia, pero probablemente el primer material de relleno comercializado y usando fase reversa fue el desarrollado en 1972 por Kirkland en Dupont. Este material se llamó Permaphase ODS, era de tipo pelicular y estaba constituido por microesferas de 35 um de diámetro con una fina capa estacionaria de silicagel unida a octadecilsilano, totalizando una partícula esférica de unos 50 um de diámetro. Antes de esto, en 1966-1967 Csaba Horvath notó que lapresión de trabajo necesaria era mayor de 1000 psi, y pensando que esta tecnología merecía un nuevo nombre fue el primero en usar las cuatro letras que hoy conocemos, HPLC(21).
En 1972, Majors desarrollo en Varian el material microparticular de 10 μm de silicagel unida a ODS. Este material se llamó MicroPak-CH. Muy poco tiempo después, en 1973 Csaba Horvath bautiza el método como Fase Reversa.
Retrospectivamente, hoy puede llamar la atención la relativamente escasa difusión que logro la cromatografía de partición liquido-liquido, aunque podemos ver que causas importantes para esto fueron debidas a la calidad del soporte, todavía no diseñado para estos propósitos, la dificultad en mantener estable la fase estacionaria en el soporte sólido y la preparación de la fase móvil. Probablemente por esta causa IUPAC ha publicado en 1972 un artículo indicando que "la fase reversa es solo una técnica de interés histórico".Sin embargo, la unión covalente de un líquido al solido de soporte fue otro hecho fundamental que llevo al desarrollo de modernas fases estacionarias, de características físicas de un sólido, pero conteniendo un líquido de interfase con unión estable.
Podemos decir que a comienzos de la década del '70 nace la HPLC, con el desarrollo de columnas rellenas con macropartículas eficientes y sistemas que permitían la elución de la fase móvil por empleo de bombas y alta presión. Como se indica más arriba, las primeras partículas utilizadas eran de tipo pelicular, consistentes en pequeñas esferas de vidrio de unos 35 μm recubiertas de una fina capa de silicagel unida covalentemente a octadecilsilano (ODS o C18), resultando en una partícula de unos 50 μm.
Poco tiempo después se desarrollaron macropartículas totalmente porosas de 10 μm de diámetro, y se fue diversificando la oferta de fases estacionarias de diferente funcionalidad, pero esto ya será desarrollado con mayor detalle en los capítulos siguientes.
Referencias
1. Tatiana A. Maryutina et al., Terminology of separation methods (IUPAC Recommendations 2017), Pure Appl. Chem.2018; 90 (1): 181-231.
2.United States Pharmacopeia. USP 41-NF 36. Rockville, MD:United States Pharmacopeial Convention;2018.
3. Nomenclature for chromatography.L.S.Ettre, Pure & Appl,Chem., Vol.65,No. 4, pp. 819-872, 1993.
4.Standard definitions of terms relating to mass spectrometry,K Murray et al., IUPAC, Anal. Chem. División, MS Terms Third Draft, August 2006.
5. Nomenclature in evaluation of Analytical methods including detec-tion and quantification capabilities, L Currie, Pure &Appl.Chem., Vol.67,No.10, pp.1699-1723,1995.
6. United States Pharmacopeial Forum 43 (5), Harmonization Stage 4: <621> Chromatography,Rockville, MD:United States Pharmaco-peial Convention US, September 2017.
7. United States Pharmacopeia <1224> Verification of Analytical Pro-cedures, USP 41-NF 36. Rockville,MD:United States Pharmacopeial Convention;2018.
8.ISO/IEC 17025:2017,General requirements for the competence of testing and calibration laboratories, section 5.4.2.
9. CFR 211.194 (a)(2),Laboratory Records.
10.How to meet ISO 17025 Requirements for Method Verification. ALACC Guide 2007,AOAC International 481 N.Frederick Ave,Suite 500, Gaithersburg, MD 20877,USA.
11. Milestones in Chromatography, The Birth of Partition Chromatogra-phy,Leslie S.Ettre, LCGC 19,506-512, Number 5 MAY 2001.
12.Csaba Horvath and the Development of the First Modern High-Performance Liquid Chromatograph, L.Ettree,LCGC North America, May 01,2005.
13. Developments in HPLC Column Packing Design,Ronald Majors, LCGC,LC column technology supplement pp 8-15, APRIL 2006.
14. The History of Chromatography, The early days of HPLC at Dupont, R.Henry,LCGC North America, Volume 27, Number 2, pp 146-153, February 2009.
15. History of Chromatography, Joseph Jack Kirkland: HPLC particle Pioneer, LC·GC Europe, pp. 438, 443,August 2013.
16. Chapters in the Evolution of Chromatography, L. Ettree, ed. By J. Hinshaw,Imperial College Press,2008,World Scientific Publishing Co.Pte.Ltd.
17.HPLC,Advances and Perspectives vol. 1, Ed. By Czaba Horvath, Academic press,1980.
18. Apparatus for Countercurrent Distribution,Lyman C. Craig, Otto Post, Anal. Chem. 21,500, 1949.
19. A.J.P.Martin and R.L.M.Synge,Biochem.J.35,91(1941).
20. A.J.P. Martin and R.L.M.Synge,Biochem.J.35,1358-1368(1941).
21.A.J.P.Martin and Howard,Biochem.J.46,532(1950)-LL partition paraffin oil and n-octane on diatomaceous earth.