
Grundlagen der Flüssigchromatographie
Definition und primäre Klassifizierung der Chromatographie
Laut IUPAC (1-4) können wir das Verfahren definieren als "ein physikalisches Trennverfahren, bei dem die Bestandteile der zu trennenden Probe auf zwei Phasen verteilt werden, von denen eine stationär bleibt (stationäre Phase), während sich die andere (die mobile Phase) in einer bestimmten Richtung bewegt.
Die stationäre Phase ist eine der beiden Phasen, aus denen das chromatographische System besteht. Sie kann ein Feststoff, ein Gel oder eine Flüssigkeit sein. Wenn es sich um eine Flüssigkeit handelt, kann sie über einen Feststoff verteilt werden...
Die mobile Phase ist eine Flüssigkeit, die in einer bestimmten Richtung durch oder entlang eines stationären Bettes perkoliert. Sie kann eine Flüssigkeit (Flüssigkeitschromatographie) oder ein Gas (Gaschromatographie) sein..
Nach der IUPAC kann die Chromatographie auf verschiedene Weise klassifiziert werden, und zwar nach der Methode, der Form des stationären Bettes, der physikalischen Beschaffenheit der mobilen Phase und nach der Art der Trennung (siehe Abbildung 1.1.).
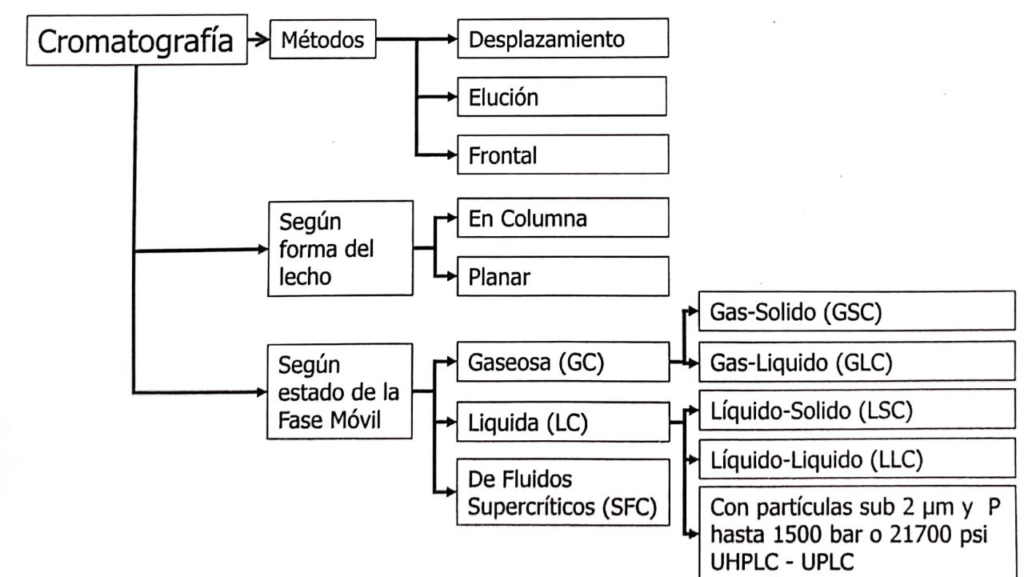
In relation to liquid chromatography (LC) on column, we can find LC with large particles operated by gravity or at low pressure, known as liquid chromatography on column, and LC with microparticles that require the use of pumps for the elution of the mobile phase. If the microparticles are larger than 2 μm it is possible to use an HPLC system, but if they are less than 2 μm it is necessary to use a UPLC or UHPLC system.
Die Klassifizierung nach dem Trennungsmechanismus ist etwas umfangreicher und komplexer und umfasst die folgenden(1).
| Method Designation | Separation mechanism |
|---|---|
| Adsorptions-Chromatographie | Unterschied in der Adsorptionsaffinität zwischen Probenbestandteilen und den aktiven Stellen der stationären Phase. aktive Stellen der stationären Phase |
| Affinitäts-Chromatographie | Biologische Spezifität der Analyt-Ligand-Interaktion |
| Gegenstrom-Chromatographie(CCC) | Teilung zwischen zwei nicht mischbaren flüssigen Phasen ohne festen Träger |
| Chirale oder enantioselektive Chromatographie | Trennung durch Wechselwirkung mit einem chiralen Partner im System, typischerweise die stationäre Phase, kann aber auch in der mobilen Phase sein. |
| Größenausschlusschromatographie (SEC) | Trennung durch Ausschluss (Größe und/oder Form des Analyten) mit Kalibrierungskurven auf der Grundlage des Molekulargewichts. Je nach mobiler Phase kann es sich um GFC (wässrig) oder GPC (organisch) handeln. |
| Hydrodynamische Chromatographie | Trennung von Makromolekülen durch den Geschwindigkeitsunterschied der mobilen Phase zwischen der Achse und der Achse und den Wänden der Säule |
| Interaction Chromatography Hydrophilic-HILIC | Trennungs-/Ionenaustausch in einer Normalphasensäule unter Verwendung einer organischen mobilen Phase mit einem geringen Anteil an Wasser. mit einem geringen Anteil an Wasser |
| Wechselwirkungschromatographie Hydrophob-HIC | Trennung durch Wechselwirkung zwischen hydrophoben Stellen der stationären Phase und hydrophoben Bereichen der Proteine, die durch die Wirkung der Ionenstärke freigelegt werden |
| Ionenaustauschchromatographie | Trennung durch Ionenaustausch zwischen funktionellen Gruppen der Säule und Probenbestandteilen. Der Austausch kann von Anionen oder Kationen erfolgen. |
| Ionenausschlusschromatographie | Partikelausschluss und frühzeitige Elution von ionischen Komponenten, während entgegengesetzt geladene Verbindungen zurückgehalten und anschließend eluiert werden |
| Ionen-Paarungschromatographie | Aufteilung der Ionenpaare, die sich zwischen dem ionischen Analyten und einem Zusatzstoff mit entgegengesetzter Ladung in einem geeigneten Puffer bilden. entgegengesetzte Ladung, in einem geeigneten Puffer |
| Ligandenaustauschchromatographie | Trennung auf der Grundlage der Fähigkeit des Analyten, mit Metallen Komplexe zu bilden |
| Mizellare Chromatographie | Umkehrphasenchromatographie, bei der die mobile Phase ein Tensid in einer Konzentration enthält, die höher ist als die kritische Mizellenkonzentration |
| Normalphasen-Chromatographie | Trennung, bei der die stationäre Phase stärker polar ist als die mobile Phase |
| Trennungs-Chromatographie | Trennung aufgrund der unterschiedlichen Löslichkeit der Probenbestandteile zwischen der mobilen und der stationären Phase. zwischen der mobilen Phase und der stationären Phase |
| Umkehrphasen-Chromatographie | LC-Trennung, bei der die mobile Phase stärker polar ist als die stationäre Phase |
HPLC - UPLC - UHPLC
HPLC stands for High Performance Liquid Chromatography, which will be translated here as High Performance Liquid Chromatography, keeping that acronym. Diese Bezeichnung wird anderen, in anderen Medien verwendeten Bezeichnungen wie CLAP, High Performance oder High Efficiency vorgezogen, die auch von anderen Quellen verwendet werden. Sie wird auch nicht als Hochdruck-Flüssigkeitschromatographie übersetzt, in Anlehnung an das erstmals von Csaba Horvath, dem Pionier dieser Wissenschaft, verwendete Akronym, und zwar aus dem einfachen Grund, dass Druck kein Trennwerkzeug ist, sondern die natürliche Folge des erzwungenen Durchgangs einer Flüssigkeit durch eine mit Makropartikeln gefüllte Säule. Eine klarere Tatsache, die jedoch zu einigen Missverständnissen geführt hat, ist das Aufkommen der UHPLC. Da "herkömmliche" HPLC-Systeme mittlerweile Drücke bis zu 6000 psi erzeugen können, wurde dieser Druck zum Industriestandard. Die Entwicklung der Partikelchromatographie zu Durchmessern von weniger als 2 μm machte es jedoch erforderlich, diese Barriere zu überwinden. Der erste, der dies tat, war die Waters Corp., die das UPLC®-System "Ultra Performance Liquid Chromatography" nannte und den Namen und das Akronym markenrechtlich schützen ließ, wodurch die Verwendung dieses Akronyms durch andere Gerätehersteller verhindert wurde.
Diese Systeme können heute Drücke von bis zu 1500 atm oder fast 22000 psi erreichen, und es ist klar, dass das Ziel ihrer Entwicklung einfach darin bestand, die geschaffenen Sub-2-Partikel zu nutzen, wofür es notwendig war, den maximalen Arbeitsdruck zu erhöhen und das Totvolumen des Systems so weit wie möglich zu verringern, das Zellenvolumen zu reduzieren und die Ansprechgeschwindigkeit des Detektors zu erhöhen, wesentliche Faktoren, um die mit der Verringerung der Partikelgröße gewonnene Effizienz nicht zu verlieren.
Da es gelang, bei höherem Druck mit Partikeln kleineren Durchmessers und damit effizienter zu arbeiten, wurde diese Methode offenbar als besser und damit anders als die HPLC angesehen, die einen anderen Namen verdiente. Angesichts der Neuheit lehnte die kommerziell benachteiligte Konkurrenz dieses Verfahren zunächst mit dem Argument ab, dass es sich nicht lohne, bei so hohem Druck zu arbeiten, was die Vorstellung verstärkte, dass es sich um ein anderes System handele. Mit zunehmender Verbreitung der Methode und angesichts der höheren Arbeitsgeschwindigkeit, der größeren Effizienz und des geringeren Lösungsmittelverbrauchs begannen schließlich die Entwicklung und die allgemeine Produktion dieser Geräte, die als UHPLC bezeichnet wurden, um die Verwendung eines bereits eingetragenen Namens zu vermeiden.
Die Folge war, dass es in regulierten Bereichen, z. B. USP, EP, JP, nicht möglich war, Verfahren, die in der HPLC entwickelt und validiert wurden, auf die UHPLC zu übertragen, einfach weil es sich um eine andere Trennmethode handeln würde. Die Säulen, Füllstoffe und die Funktionalität der Füllstoffe sind jedoch die gleichen, die mobile Phase ist die gleiche, die Trennmechanismen, die Interpretation der Chromatogramme und die Berechnungen sind die gleichen. Es musste abgewartet werden, bis das System ausgereift war und die Gleichwertigkeit experimentell nachgewiesen werden konnte, um eine Übertragung der Verfahren zu ermöglichen, was 2017 zum ersten harmonisierten Dokument des Chromatographie-Kapitels der USP (5), EP und JP führte, in dem die Übertragung von Methoden von der HPLC auf die UHPLC und umgekehrt in regulierten Umgebungen vorgeschlagen wurde. Dies ändert natürlich nichts daran, dass offizielle Methoden vor der Anwendung verifiziert werden sollten, wie von USP (6), FDA (7), ISO(8) und AOAC (9) vorgeschrieben, unabhängig davon, ob sie auf UHPLC übertragen wurden oder nicht. Das wichtigste Konzept ist die Schlussfolgerung, dass beide Modalitäten gleichwertig sind und dass offizielle Verfahren ohne Verlust des "offiziellen" Status aufgestockt, verifiziert und wie ursprünglich validiert verwendet werden können.
Der Mechanismus der Trennung
Um auf die IUPAC-Definition zurückzukommen: "Die Chromatographie ist ein physikalisches Trennverfahren, bei dem die Bestandteile der zu trennenden Probe in zwei Phasen verteilt sind...".
Wir müssen also ein Drei-Komponenten-System bewerten: die Probe, die stationäre Phase und die mobile Phase. In Abbildung 1.2 besteht die Probe aus drei Komponenten, von denen jede eine andere chemische Struktur hat, die durch das Vorhandensein von funktionellen Gruppen gekennzeichnet ist, die sie identifizieren, z. B. phenolische oder alkoholische Hydroxylgruppen, primäre oder sekundäre Amine, Aldehyde, Ketone usw. Die mobile Phase und die stationäre Phase haben wiederum ihre eigenen funktionellen Gruppen, die ihnen eine Affinität für komplementäre funktionelle Gruppen verleihen (siehe Abbildung 1.1).
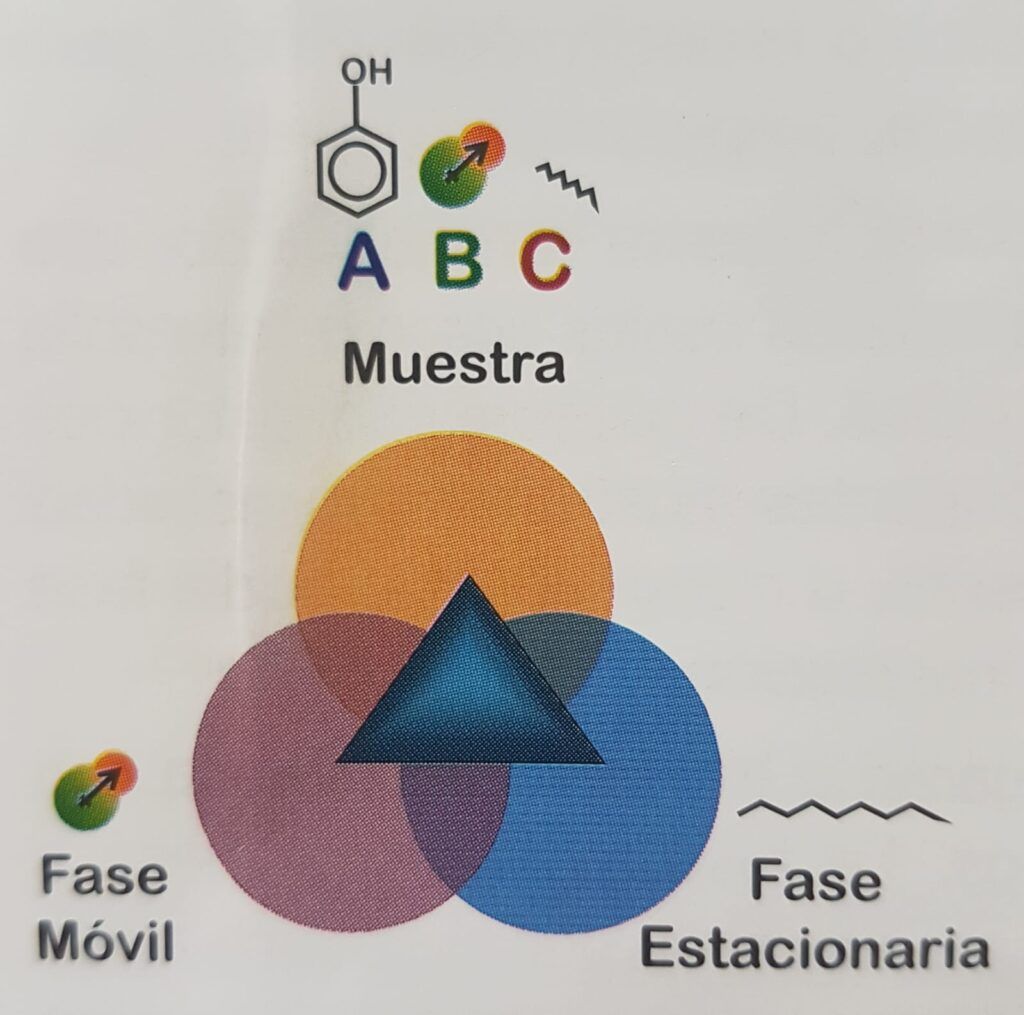
Es gibt mehrere Wechselwirkungen, die die Affinität zwischen Molekülen modellieren. Eine der wichtigsten ist die Wasserstoffbrücke, bei der ein Wasserstoffatom kovalent an ein elektronegatives Atom in einem protonenabgebenden Molekül gebunden ist, das es freilegt. In der Umgebung kann es ein Protonenakzeptormolekül mit einem anderen elektronegativen Atom finden, das sich nach dem Proton sehnt, das es anzieht, es aber nicht nehmen kann, weil es kovalent an das erste Molekül gebunden ist. Diese Wechselwirkung ist in der Natur weit verbreitet, z. B. zwischen Carbonyl- und Aminogruppen in Proteinen zur Bildung ihrer dreidimensionalen Struktur, zwischen komplementären Basen der DNA, zwischen Wasser und dem Hydroxyl eines Alkohols oder einem Carbonyl usw.
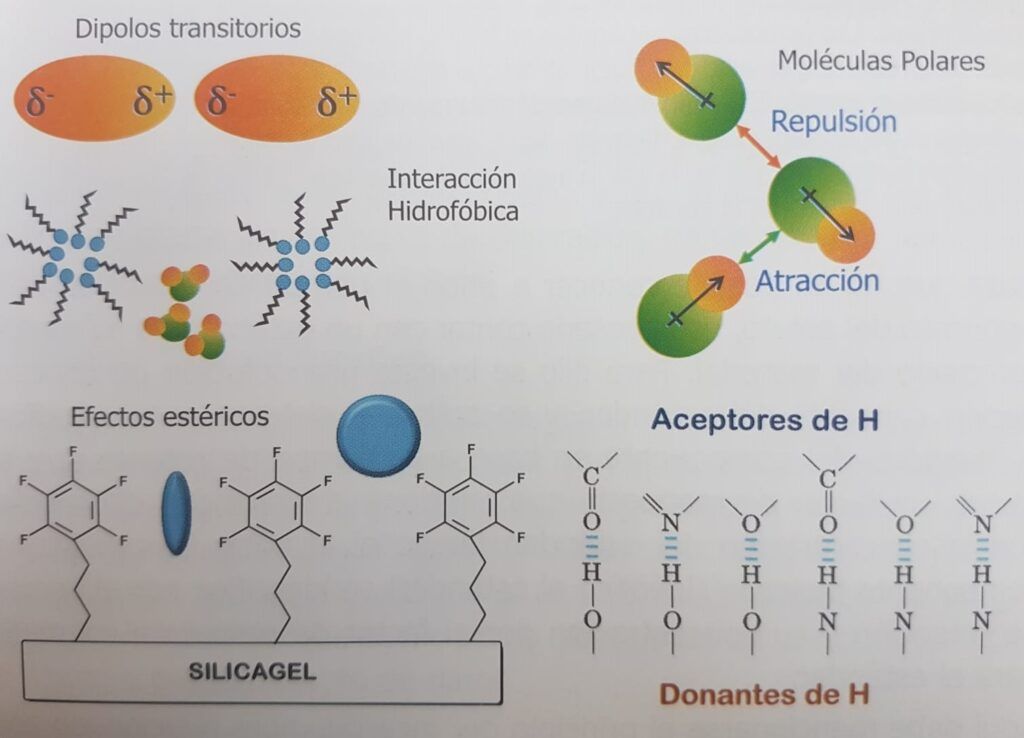
Zu den weiteren Wechselwirkungen gehören elektrostatische Wechselwirkungen zwischen Anionen und Kationen, hydrophobe und hydrophile Wechselwirkungen, die die Vermischung von Öl und Wasser, die zusammenklumpen, erschweren, sterische Wechselwirkungen, die es einem Molekül erschweren, in die gebundene Phase einzutreten, und zwar aufgrund sterischer Hindernisse, die einfach auf die Größe des Analyten zurückzuführen sind, die schwachen Londoner Kräfte, die durch die vorübergehenden Dipole entstehen, die in jedem Atom durch die Bewegung seiner Elektronen erzeugt werden, die Dipol-Wechselwirkungen, die durch Moleküle mit permanenter Polarität wie Acetonitril entstehen, sowie andere Retentionskräfte (Abbildung 1.3)..
Bei der chromatographischen Trennung (Abbildung 1.4) wird die in einer Säule enthaltene stationäre Phase mit einem konstanten Strom mobiler Phase äquilibriert. Der Säulenausgang ist mit einem Überwachungsgerät verbunden, das eine physikalisch-chemische Eigenschaft der eluierenden Flüssigkeit misst, im Falle der Abbildung ein photometrischer oder spektrophotometrischer Detektor, es kann sich aber auch um einen Leitfähigkeits-, Fluoreszenz- oder amperometrischen Detektor usw. handeln.
Sobald das System im Gleichgewicht ist, wird die Probe injiziert, die aus einer Lösung mit verschiedenen Komponenten besteht, die durch die Säule transportiert werden. Je nach chemischer Funktionalität weist jede der Komponenten eine höhere Affinität für die mobile Phase auf und eluiert schneller, oder eine höhere Affinität für die stationäre Phase und wird entsprechend dieser Wechselwirkung zurückgehalten.
Infolgedessen durchlaufen die Komponenten die Säule mit unterschiedlichen Geschwindigkeiten und werden getrennt. Die eluierte mobile Phase durchläuft eine Durchflusszelle, wo sie von dem entsprechenden System überwacht wird, in diesem Fall von einem Lichtstrahl, der durch die Durchflusszelle zu einer Fotozelle geleitet wird, die auf die Veränderung der Lichtdurchlässigkeit/Absorption reagiert. Die mobile Phase setzt ihren Weg fort, und das Signal der Fotozelle, das proportional zur momentanen Absorption des Eluats ist, das die Zelle durchläuft, wird an einen Schreiber oder an ein Datenaufzeichnungs- und -verarbeitungssystem auf einem Computer übertragen. Das Ergebnis ist ein Chromatogramm, eine Darstellung des von der Fotozelle erzeugten Signals als Funktion der Zeit (siehe Abbildungen 1.7 a und b).
Das Chromatogramm zeigt dann die Trennung der Komponenten der eingespritzten Lösung in einem Diagramm des vom Detektor erfassten Signals (mV) als Funktion der Zeit (Minuten). Die Retentionszeit (als Peak-Apex-Elutionszeit) ist eine Folge der chemischen Struktur und der Funktionalität und daher eine primäre Funktion der Identität der Komponente. Die Retentionszeit von A identifiziert in erster Linie die Verbindung A als Bestandteil der eingespritzten Lösung, ebenso wie B und C. Die Intensität des Signals, gemessen als Peakhöhe oder Fläche unter der Kurve, ist eine primäre Funktion der Konzentration.

Da es nicht möglich ist, die Retentionszeit oder das Ansprechverhalten des gelösten Stoffes a priori zu kennen, ist es notwendig, einen geeigneten Referenzstandard des Materials zu haben. Zu diesem Zweck wird eine Lösung mit bekannter Konzentration des Standards eingespritzt, und das System wird kalibriert, um die Identität der Komponente auf der Grundlage ihrer Retentionszeit zuzuordnen, und es wird ein Reaktionsfaktor berechnet, der die Intensität des Signals mit der Konzentration des Standards in Beziehung setzt. Wenn dann die Probe eingespritzt wird, wird die (mit dem Standard identische) Zielkomponente anhand der Retentionszeit und ihre Konzentration anhand des für den Standard geschätzten Reaktionsfaktors identifiziert.
An dieser Stelle sollte das Unschärfeprinzip in Bezug auf die Identität erwähnt werden. Es ist zwar klar, dass die Identität der Komponenten A, B und C von der Retentionszeit abhängt, aber es ist auch klar, dass es in der Natur sicherlich Dutzende oder Hunderte von Verbindungen gibt, die mit einer Retentionszeit eluieren, die der Retentionszeit der Zielkomponente ähnelt oder nicht von ihr zu unterscheiden ist. Im Falle der Abbildung kann es Hunderte von Verbindungen geben, die in einem 10-minütigen Lauf eluieren. Die erste Behauptung, die Identität, ist also zuverlässiger, wenn die Variabilität der Komponenten nicht sehr groß ist, z. B. in einer Industrie, die einige Dutzend Materialien zur Herstellung einiger weniger Produkte verarbeitet. Mit zunehmender Vielfalt wird ein Bedarf an Instrumenten zur Identitätsbestätigung bestehen, z. B. durch Strukturanalyse (gleichzeitige spektroskopische Analyse, Analyse der Peak-Reinheit, Molekulargewicht), um die Gültigkeit des Ergebnisses zu kontrollieren. Und es wird Fälle geben, in denen die Vielfalt absolut komplex ist, wie bei bioanalytischen Tests, bei denen die biologischen Matrizes eine sehr hohe Anzahl potenzieller Störfaktoren aufweisen, oder bei einem Umweltscreening oder bei der Analyse einer bestimmten Struktur in natürlich komplexen Matrizes wie Früchten oder Pflanzenextrakten. In diesen Fällen kann es absolut notwendig sein, sehr selektive Detektoren zu haben, wie z. B. Masse-Masse- oder Flugzeit-Kopplungen.
In Bezug auf die konzentrationsbezogene Unsicherheit ist es klar, dass eine Korrelation zwischen der Reaktion und der Konzentration des Standards im verwendeten System gesucht werden muss und dass die Reaktion des Analyten auf dieser Kalibrierkurve in einem qualifizierten System unter Verwendung eines validierten Verfahrens interpoliert werden muss.
Das chromatographische System
Abbildung 1.5 zeigt schematisch die einfachste Version eines HPLC-Systems. Erforderlich sind mindestens ein Reservoir für die mobile Phase, eine Pumpe, ein Injektor, eine Säule und ein Detektor mit den dazugehörigen Schläuchen und Verbindungen. Der Detektor sollte mindestens an einen XY-Schreiber angeschlossen sein, obwohl die moderne Analytik die Verwendung eines Computers als Datenerfassungs-, -speicherungs- und -verarbeitungssystem empfiehlt.

Obwohl dieses Grundschema für eine Vielzahl von Anwendungen ausreichend sein mag, ist seine Ausweitung auf komplexere Systeme aufgrund des Optimierungs- und Automatisierungsbedarfs unvermeidlich (siehe Abbildung 1.6). So hängt beispielsweise die Zuverlässigkeit der instrumentellen Analyse in hohem Maße von der statistischen Analyse ab, die die Verarbeitung von Wiederholungen zur Berechnung der Unsicherheit fördert, was dazu führt, dass automatische Injektoren erwünscht sind, die ihrerseits eine Optimierung von Zeit und Ressourcen ermöglichen.
Dann ist es manchmal notwendig, Elutionsgradienten zu verwenden, was die Verwendung mehrerer Lösungsmittel zur Vorbereitung der mobilen Phase in situ ermöglicht, die durch Online-Entgaser laufen, bevor sie die Pumpe erreichen. Der Gradient kann mit einer oder zwei Pumpen gebildet werden.
Es ist dann möglich und manchmal auch praktisch, einen Säulenofen zu verwenden, um eine oder mehrere Chromatographiesäulen aufzunehmen. Das System kann so programmiert werden, dass die erste Säule verwendet, gewaschen und konditioniert wird, die zweite Säule verwendet und gewaschen wird und das System nach Beendigung abgeschaltet wird, ohne dass ein Analytiker tätig werden muss. Die Vielfalt der Analyten kann die Verwendung von mehr als einem Detektor erforderlich machen, und das Säuleneluat muss nicht unbedingt verworfen werden, sondern die einzelnen Komponenten können mit geeigneten Systemen gesammelt werden.

| Chromatogram | System suitability test |
|
| Material | Standard solution 1018 | Source: QA Laboratory |
| Analyst | FQ-SR | Vial: 1 |
| Date: | 07/26/2022 | Time: 12:19:46 |



Kurze Geschichte der Flüssigchromatographie
Im Folgenden wird die geschichtliche Entwicklung der Flüssigchromatographie und der HPLC anhand von Artikeln namhafter Chromatographieforscher skizziert. Es ist nicht beabsichtigt, eine erschöpfende oder vollständige Beschreibung der Entwicklung der Chromatographie zu geben und ist sicherlich unfair gegenüber vielen Forschern, deren Beitrag grundlegend für diese wesentliche Technik in praktisch allen modernen Wissenschaften war, aber die Lektüre kann helfen, die Mechanismen zu verstehen und vielleicht die aktuellen Verfahren zu verbessern. Viele der zitierten Ereignisse mögen kurz den Ursprung und einige Meilensteine aufzeigen, die es uns ermöglicht haben, hierher zu gelangen(11-17).
Am Anfang steht die Erfindung der Chromatographie, die wir Mikhail Semyonovich Tsweet verdanken, der sie im Jahr 1900 entwickelte. Zweet wurde 1872 in Asti, Italien, als Sohn einer italienischen Mutter und eines russischen Offiziers geboren. In jungen Jahren zog er in die Schweiz, wo er ein Studium der Botanik aufnahm. Er folgte seinem Vater, als dieser nach Russland zurückgerufen wurde, und ließ sich in St. Petersburg nieder. Er musste seine Abschlüsse in Moskau, wo er akademische Tätigkeiten ausübte, sowie in Warschau und Litauen revalidieren. Die Bezeichnung Chromatographie verwendete er erstmals 1906 in seinen Veröffentlichungen (siehe Abbildung 1.8). Bei seinen Arbeiten füllte er Glasröhren mit einem Feststoff, im Falle der Abbildung mit Kalziumkarbonat, und brachte sie mit einer Flüssigkeit, Schwefelkohlenstoff, ins Gleichgewicht. Nachdem er am oberen Ende Chlorophylle in Lösung eingebracht hatte, eluierte er die Flüssigkeit durch die Säule, woraufhin sich die Pigmente in der Säule trennten und eine Reihe farbiger Banden zum Vorschein kamen. Damals schuf er den Namen Chromatographie, der sich aus dem griechischen chroma (Farbe) und grapho (Schrift) zusammensetzt, d. h. Schrift in Farbe.

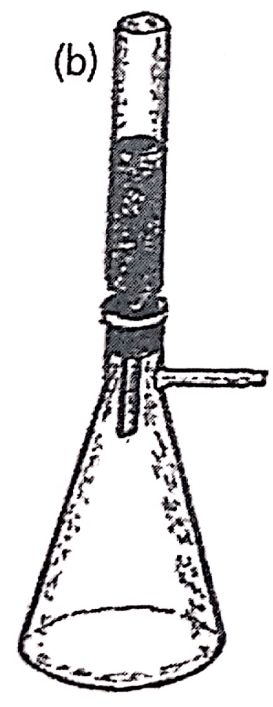

Abbildung 1.8. Zeichnungen des 1906 von Tsweet veröffentlichten Artikels. Er zeigt (a) einen Apparat mit fünf gleichzeitigen Säulen mit einem Quecksilbermanometer und einem Gummiball für die Abgabe der mobilen Phase. Sie zeigt außerdem (b) eine Trichtersäule mit einem Durchmesser von 2-3 mm und einer Länge von 20-30 cm, die auf einem Kitasat montiert ist, und (c) eine chromatographische Trennung von Pigmenten unter Verwendung von Kohlenstoffcarbonat als stationärer Phase und Schwefelkohlenstoff als mobiler Phase.Reproduziert mit Genehmigung von LC-GC.
In Wirklichkeit wurden die Probenbestandteile nicht von der Säule eluiert, wie wir es heute tun, sondern bildeten ein Farbmuster innerhalb der Säule, das die Zusammensetzung der Chlorophylle darstellt. Die Methode hatte große Auswirkungen, aber natürlich auch große Einschränkungen, da sie farblose Komponenten nicht sichtbar machen konnte und die Elution der Komponenten der Säule nicht zuließ.
Die Dünnschichtchromatographie (TLC) bietet eine Lösung für dieses Problem: Die TLC kann als offene Säulenchromatographie betrachtet werden, so dass durch den Zugriff auf das Material je nach Bedarf eine spezifische oder universelle Entwicklung durchgeführt werden kann. Die Anwendung eines Ninhydrinsprays und die anschließende Erhitzung machen beispielsweise die farblosen Aminosäuren sichtbar, die sich blau färben. Joddämpfe, denen die Trockenplatte nach dem Durchlauf ausgesetzt wird, fixieren organische Verbindungen mit Doppel- oder Dreifachbindungen und färben sie rötlich-braun. Durch einfaches Besprühen mit Schwefelsäure und anschließendes Erhitzen im Ofen bei 100°℃ werden die organischen Verbindungen verkohlt, was als dunkle Flecken auf der weißen Platte sichtbar wird.
HPTLC von Kamillenöl, verschiedene Offenbarungen
UV 254 nm mit und ohne Filter
UV 366 nm Nachderivatisierung
Anisaldehyd Drifting Reflactance R+T Transm.

Einer der ersten Wegbereiter dieser Technik war Edgar Lederer, und in jüngerer Zeit wurde sie von Egon Stahl weiterentwickelt. Die Wirkung der TLC war unmittelbar, und aufgrund ihrer Einfachheit und geringen Kosten setzte sie sich schnell in der analytischen Chemie durch. Während sich die Flüssig- und Gaschromatographie weiter verbreitete, entwickelte sich die TLC in Osteuropa weiter. Sie wird auch heute noch verwendet, wenngleich ihre Verbreitung durch den Vormarsch der HPLC stark zurückgegangen ist.
Kurz darauf erlebte die Flüssigkeitschromatographie (LC) in den 1940er Jahren einen bedeutenden Durchbruch, als Archer J.P. Martin und Richard L.M. Synge die Partitionschromatographie oder Flüssig-Flüssig-Chromatographie (LLC) im Gegensatz zur damals vorherrschenden Flüssig-Fest-Chromatographie (LSC) schufen, und diese Entwicklung war so einflussreich, dass sie 1952 den Nobelpreis für Chemie dafür erhielten.
Martin forschte im Dunn Nutritional Laboratory an Vitamin E und experimentierte mit der Trennung heterogener Phasen von Carotinen. Zu diesem Zweck hatte er ein System aus 45 aneinandergereihten Röhren konstruiert, die als Umfüllampullen dienten und durch 90 Ventile getrennt waren, die den Rückfluss der Flüssigkeit in die vorherige Ampulle verhinderten. Das Schema seiner Apparatur wurde nicht veröffentlicht.
Synge seinerseits untersuchte die Aminosäuren der Wolle im IWS, und die angewandte Methode bestand darin, die Aminosäuren zu acetylieren, um sie in Chloroform zu extrahieren. Er stellte mit Begeisterung den Unterschied in den Chloroform-Wasser-Verteilungskoeffizienten der acetylierten Aminosäuren fest, was vielversprechende Aussichten für die Flüssig-Flüssig-Extraktion bot, und wurde dafür von Martin kontaktiert, der sehr erfahren in der Gegenstromverteilung war und eine Apparatur entwickelt hatte, die ihn in Cambridge bekannt gemacht hatte. Kurze Zeit später arbeiteten sie bei der Wool Research Association in Leeds, UK, wo sie eine weitere Apparatur entwickeln mussten, da die vorhandene nicht mit dem Wasser-Chloroform-System kompatibel war. Der neue Durchlaufapparat hatte 39 "theoretische Platten" (Abbildung 1.10 zeigt ein von Lyman Craig in den 40er Jahren entwickeltes System (18), das nicht unbedingt mit Martins System identisch ist, aber einen Gegenstromextraktionszug zeigt, der in jenen Jahren verwendet wurde).
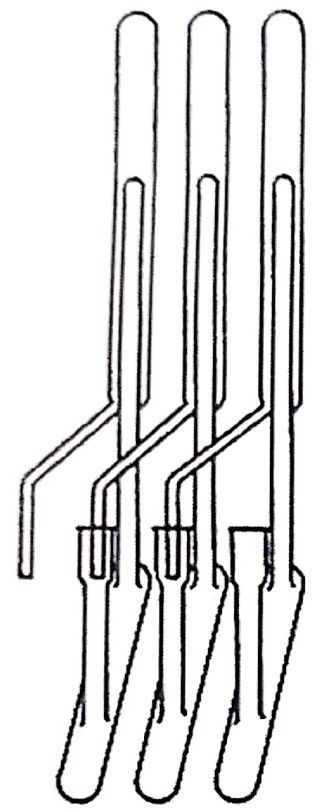

Berichten zufolge handelte es sich um ein kompliziertes System, das von zwei Bedienern im 4-Stunden-Takt bedient werden musste, die durch Chloroformdämpfe in den Schlaf gewiegt wurden. Die Ergebnisse waren nicht sehr zufriedenstellend, und Martin und Synge versuchten, das System durch die Entwicklung von Varianten zu verbessern. Ein Zwischensystem war ein Glasrohr, bei dem die Wolle und die Baumwollfasern in Richtung des Rohrs ausgerichtet waren, wobei sich das Chloroform in eine Richtung und das Wasser in die entgegengesetzte Richtung bewegte. Beides funktionierte nicht wie erwartet, bis Martin schließlich auf die Idee kam, dass es nicht notwendig sei, die beiden Flüssigkeiten zu bewegen, dass man eine in einer Säule stehen lassen und die andere bewegen könne.
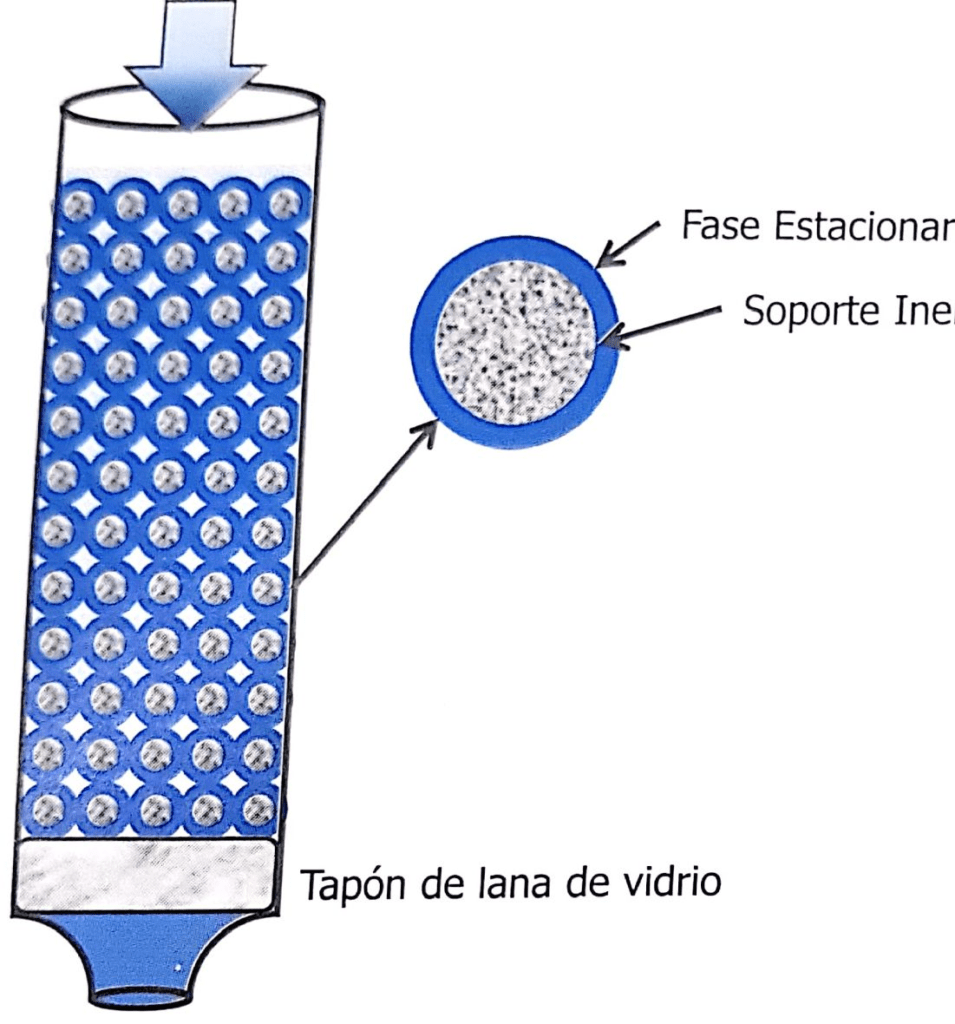
Zu diesem Zweck imprägnierten sie das Kieselgel, das sie in der Waage als Trockenmittel verwendeten, mit Wasser und füllten mit diesem Material eine Glassäule, fügten das acetylierte Aminosäuregemisch hinzu und gossen Chloroform in den Einlass, etwa wie in der schematischen Darstellung in Abbildung 1.11). Durch die Zugabe von Methylorange konnten sie den Weg der Aminosäuren durch die Säule verfolgen. Das mag heute sehr einfach klingen, aber es war sicherlich harte Arbeit, die stationäre Phase (Wasser) auf einem Träger, der nicht für diesen Zweck hergestellt worden war (Kieselgel), und die mobile Phase (Chloroform) in reproduzierbarer Weise richtig vorzubereiten. Martin entwickelte parallel dazu die Theorie der Chromatographie auf der Grundlage von Destillationskonzepten, und die Arbeit wurde in zwei Teilen im November 1941 dem Biochemical Journal vorgelegt und im Dezember 1941 als A New Form of Chromatography Employing Two Liquid Phases (19-20) veröffentlicht.
Kurz darauf verließ Synge die Gruppe, um Aminosäuren in Antibiotika zu untersuchen, und war der erste, der die Sequenz in einem Polypeptid mit Hilfe der Papierchromatographie aufklärte.
Martin trat 1948 dem Medical Research Council bei und beschäftigte sich mit der Analyse von Fettsäuren durch Chromatographie. Da sein System über eine polare stationäre Phase und eine wenig polare mobile Phase (Wasser und Chloroform) verfügte, waren die Verteilungskoeffizienten nicht günstig, so dass er beschloss, eine hydrophobe stationäre Phase zu entwickeln, um die Fettsäuren zurückzuhalten. Dies gelang ihm mit Howard (20) durch die Behandlung von Kieselgur mit Dimethyl-Dichlorsilan (siehe Abbildung 1.12).
Im Gegensatz zur '41 entwickelten Flüssig-Flüssig-Trennchromatographie, bei der die Bindung der Flüssigkeit an die stationäre Phase eine einfache mechanische Retention in den Poren war und daher instabil, band diese Reaktion das silanisierende Molekül durch kovalente Bindung an den Träger, so dass die unpolare Flüssigkeit (Dimethylsilan) als hydrophobe stationäre Phase übrig blieb, die mit dem polaren Lösungsmittel der mobilen Phase nicht mischbar war. Martin verwendete dieses System zur Trennung von Laurinsäure und Stearinsäure durch Partitionierung.

Andererseits hatte Martin 1941 die Verwendung einer gasförmigen mobilen Phase vorausgesagt, entwickelte die Idee aber erst 1950 weiter, als er die silanisierte stationäre Phase in die GC einführte und die Methode im Oktober 1950 als Gas-Flüssigkeits-Verteilungschromatographie veröffentlichte. Diese Methode erwies sich in kurzer Zeit als nützlich und bildete die Grundlage für die Entwicklung der Gas-Flüssig-Chromatographie (GLC).
Innerhalb kurzer Zeit wanderte dieses Material zurück in die Flüssigchromatographie und wurde erstmals 1967 von Aue und Hasting an der Dalhouise University in Nova Scotia eingesetzt. Das wahrscheinlich erste kommerziell genutzte Füllmaterial mit Umkehrphase wurde jedoch 1972 von Kirkland bei Dupont entwickelt. Dieses Material wurde Permaphase ODS genannt, war vom pellikulären Typ und bestand aus Mikrokügelchen mit einem Durchmesser von 35 um mit einer dünnen stationären Schicht aus Kieselgel, die an Octadecylsilan gebunden war, so dass insgesamt ein kugelförmiges Teilchen mit einem Durchmesser von etwa 50 um entstand. Zuvor, in den Jahren 1966-1967, stellte Csaba Horvath fest, dass der benötigte Arbeitsdruck mehr als 1000 psi betrug, und da er der Meinung war, dass diese Technologie einen neuen Namen verdiente, verwendete er als erster die vier Buchstaben, die wir heute kennen: HPLC(21).
1972 entwickelte Majors bei Varian das Mikropartikelmaterial aus 10 μm Kieselgel, das an ODS gebunden ist. Dieses Material wurde MicroPak-CH genannt. Kurz darauf, 1973, taufte Csaba Horvath die Methode auf den Namen Reverse Phase.
Rückblickend mag die relativ geringe Verbreitung der Flüssig-Flüssig-Trennchromatographie heute auffallen, obwohl wir erkennen können, dass wichtige Ursachen dafür in der Qualität des Trägers, der noch nicht für diese Zwecke ausgelegt war, in der Schwierigkeit, die stationäre Phase auf dem festen Träger stabil zu halten, und in der Zubereitung der mobilen Phase lagen. Wahrscheinlich aus diesem Grund veröffentlichte die IUPAC 1972 einen Artikel, in dem es hieß, dass "die umgekehrte Phase nur eine Technik von historischem Interesse ist"; die kovalente Bindung einer Flüssigkeit an den Trägerfeststoff war jedoch eine weitere grundlegende Tatsache, die zur Entwicklung moderner stationärer Phasen führte, die die physikalischen Eigenschaften eines Feststoffs aufweisen, aber eine Grenzflächenflüssigkeit mit stabiler Bindung enthalten.
Man kann sagen, dass in den frühen 1970er Jahren die HPLC geboren wurde, mit der Entwicklung von Säulen, die mit effizienten Makropartikeln gefüllt waren, und Systemen, die die Elution der mobilen Phase durch den Einsatz von Pumpen und hohem Druck ermöglichten. Wie bereits erwähnt, waren die ersten verwendeten Partikel vom pellikulären Typ, bestehend aus kleinen Glaskugeln von etwa 35 μm, die mit einer dünnen Schicht Kieselgel überzogen waren, das kovalent an Octadecylsilan (ODS oder C18) gebunden war, was zu einem Partikel von etwa 50 μm führte.
Kurz darauf wurden vollständig poröse Makropartikel mit einem Durchmesser von 10 μm entwickelt, und das Angebot an stationären Phasen mit unterschiedlicher Funktionalität wurde diversifiziert, worauf jedoch auf den folgenden Seiten näher eingegangen wird.
Hier können Sie den Beitrag auf Spanisch sehen: Cromatografia
References
A. Wikipedia: https://en.wikipedia.org/wiki/High-performance_liquid_chromatography
B. Knauer: https://www.knauer.net/en/search?q=chromatography
C. Kromasil: https://www.kromasil.com/support/faq.php
D. Shimadzu: https://www.shimadzu.com/an/service-support/technical-support/analysis-basics/basic/what_is_hplc.html
E. ChemistryView: https://www.chemistryviews.org/details/education/9464911/What_is_HPLC/
1. Tatiana A. Maryutina et al., Terminology of separation methods (IUPAC Recommendations 2017), Pure Appl. Chem.2018; 90 (1): 181-231.
2.United States Pharmacopeia. USP 41-NF 36. Rockville, MD:United States Pharmacopeial Convention;2018.
3. Nomenclature for chromatography.L.S.Ettre, Pure & Appl,Chem., Vol.65,No. 4, pp. 819-872, 1993.
4.Standard definitions of terms relating to mass spectrometry,K Murray et al., IUPAC, Anal. Chem. División, MS Terms Third Draft, August 2006.
5. Nomenclature in evaluation of Analytical methods including detec-tion and quantification capabilities, L Currie, Pure &Appl.Chem., Vol.67,No.10, pp.1699-1723,1995.
6. United States Pharmacopeial Forum 43 (5), Harmonization Stage 4: <621> Chromatography,Rockville, MD:United States Pharmaco-peial Convention US, September 2017.
7. United States Pharmacopeia <1224> Verification of Analytical Pro-cedures, USP 41-NF 36. Rockville,MD:United States Pharmacopeial Convention;2018.
8.ISO/IEC 17025:2017,General requirements for the competence of testing and calibration laboratories, section 5.4.2.
9. CFR 211.194 (a)(2),Laboratory Records.
10.How to meet ISO 17025 Requirements for Method Verification. ALACC Guide 2007,AOAC International 481 N.Frederick Ave,Suite 500, Gaithersburg, MD 20877,USA.
11. Milestones in Chromatography, The Birth of Partition Chromatogra-phy,Leslie S.Ettre, LCGC 19,506-512, Number 5 MAY 2001.
12.Csaba Horvath and the Development of the First Modern High-Performance Liquid Chromatograph, L.Ettree,LCGC North America, May 01,2005.
13. Developments in HPLC Column Packing Design,Ronald Majors, LCGC,LC column technology supplement pp 8-15, APRIL 2006.
14. The History of Chromatography, The early days of HPLC at Dupont, R.Henry,LCGC North America, Volume 27, Number 2, pp 146-153, February 2009.
15. History of Chromatography, Joseph Jack Kirkland: HPLC particle Pioneer, LC·GC Europe, pp. 438, 443,August 2013.
16. Chapters in the Evolution of Chromatography, L. Ettree, ed. By J. Hinshaw,Imperial College Press,2008,World Scientific Publishing Co.Pte.Ltd.
17.HPLC,Advances and Perspectives vol. 1, Ed. By Czaba Horvath, Academic press,1980.
18. Apparatus for Countercurrent Distribution,Lyman C. Craig, Otto Post, Anal. Chem. 21,500, 1949.
19. A.J.P.Martin and R.L.M.Synge,Biochem.J.35,91(1941).
20. A.J.P. Martin and R.L.M.Synge,Biochem.J.35,1358-1368(1941).
21.A.J.P.Martin and Howard,Biochem.J.46,532(1950)-LL partition paraffin oil and n-octane on diatomaceous earth.